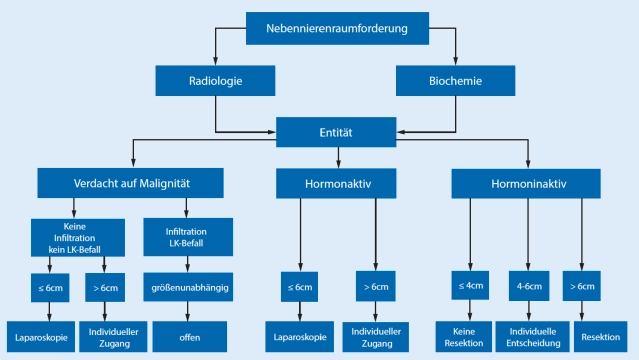
En las directrices existe consenso en que la mayoría de los tumores suprarrenales, dada la indicación, deben operarse de forma mínimamente invasiva. Diámetro del tumor >6 cm y claros indicios de malignidad en la imagen preoperatoria se consideran límites de la cirugía mínimamente invasiva.
Para la adrenalectomía abierta se deben preferir el acceso transabdominal o el toracoabdominal, ya que la indicación principal para el procedimiento abierto son tumores grandes sospechosos de malignidad.
En tumores suprarrenales sospechosos a partir de 6-8 cm de tamaño y unidades Hounsfield (HU) (valores de densidad de la TC de las suprarrenales) >20, sin prueba preoperatoria de malignidad, se debe preferir un acceso abierto (+ linfadenectomía).
Se debe aspirar a una resección R0 en bloque con el tejido adiposo retroperitoneal circundante, evitar la apertura de la cápsula tumoral, al menos los ganglios linfáticos morfológicamente llamativos en imágenes deben extirparse simultáneamente.
- Tumores endocrino-activos de la corteza suprarrenal (adenomas de Conn o Cushing, tumores con secreción de andrógenos) > 10 cm
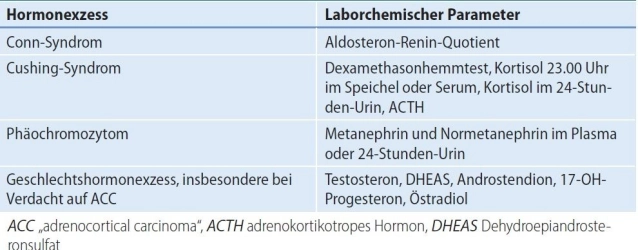
- Síndrome de Conn (hiperaldosteronismo primario, HAP):
La causa más frecuente de hipertensión secundaria es el hiperaldosteronismo primario.
En un adenoma productor de aldosterona unilateral está indicada la adrenalectomía unilateral.
En pacientes con HAP y alteraciones suprarrenales bilaterales se puede considerar una operación si el muestreo venoso suprarrenal (AVS mediante toma selectiva de sangre de venas suprarrenales) muestra una localización funcional.
- Síndrome de Cushing(hipercortisolismo)
Un síndrome de Cushing suprarrenal florido con estigmas clínicos clásicos representa una indicación quirúrgica.
Antes de una posible extirpación quirúrgica del tumor suprarrenal se debe confirmar la independencia de ACTH del exceso de cortisol, para que la intervención no se realice erróneamente, aunque la causa del exceso hormonal sea, por ejemplo, de origen hipofisario.
Exclusión de hipercortisolismo dependiente de ACTH (adenoma del lóbulo anterior de la hipófisis) (síndrome de Cushing central) o síndrome paraneoplásico con secreción ectópica de ACTH en enfermedad tumoral.
- Tumores de la corteza suprarrenal productores de hormonas sexuales
Un carcinoma adrenocortical es la causa más frecuente de una secreción patológica clínicamente relevante de andrógenos/estrógenos de la suprarrenal, los adenomas son muy raros.
- Síndrome de Conn (hiperaldosteronismo primario, HAP):
- Carcinoma de la corteza suprarrenal Carcinoma adrenocortical (CAC) Estadio ENSAT I-III Enlace a la clasificación ENSAT
- En el diagnóstico inicial casi siempre > 4cm y presenta en el 50-80% una actividad endocrina. Típica es una producción de cortisol o una producción hormonal mixta (andrógenos/estrógenos y cortisol).
- En el carcinoma de la corteza suprarrenal, la adrenalectomía abierta es el estándar de oro. En tumores < 6 cm sin indicios de infiltración local o de ganglios linfáticos (estadio ENSAT I+II) se puede realizar una adrenalectomía mínimamente invasiva.
- Se recomienda una resección tumoral radical con extirpación de la suprarrenal y de todo el tejido adiposo/conectivo en el compartimento afectado sin apertura de la cápsula y una linfadenectomía, esta en caso de datos inciertos. Hasta ahora no existe una definición respecto a la extensión de la linfadenectomía requerida.
- Nota: La tasa de recidivas locales y peritoneales está aumentada en el grupo laparoscópico según la evidencia actual. Una conversión de adrenalectomía laparoscópica a abierta empeora la supervivencia global.
- Feocromocitoma (FC) Tumor de la médula suprarrenal con exceso de catecolaminas en caso de sospecha de malignidad, tumores muy grandes o si se esperan dificultades técnicas.
- Aproximadamente 1/3 de todos los pacientes con feocromocitoma se asocian a un síndrome tumoral hereditario. El cribado genético es un componente indispensable del diagnóstico de feocromocitoma. Los tumores asociados a SDHB están asociados a un riesgo particularmente alto de malignidad y recidiva.
- En total, el 10 % de los FC son malignos. Solo las metástasis prueban un feocromocitoma maligno.
- Mielolipomas no representan per se una indicación quirúrgica.
Los mielolipomas suprarrenales grandes (tumores benignos inactivos hormonalmente que contienen tejido adiposo maduro y tejido hematopoyético) pueden volverse sintomáticos por necrosis o hemorragia espontánea. En estos casos puede ser necesaria una resección.
- Metástasis suprarrenales (de tumores malignos de otro origen) deben extirparse si no hay otras metástasis y se logra la ausencia de tumor mediante la extirpación.
Nota: La adrenalectomía por metástasis debe realizarse de forma mínimamente invasiva, siempre que la metástasis se pueda extirpar in toto y sin diseminación de células tumorales. Un procedimiento abierto se reserva para los pocos casos en los que hay indicios de infiltración local o si la metástasis supera los 6 cm.
- Grandes schwannomas o neuroblastomas adultos
- Problemas pulmonares o cardíacos, que excluyen una intervención mínimamente invasiva.
- Presencia de adherencias intraabdominales o retroperitoneales después de operaciones previas