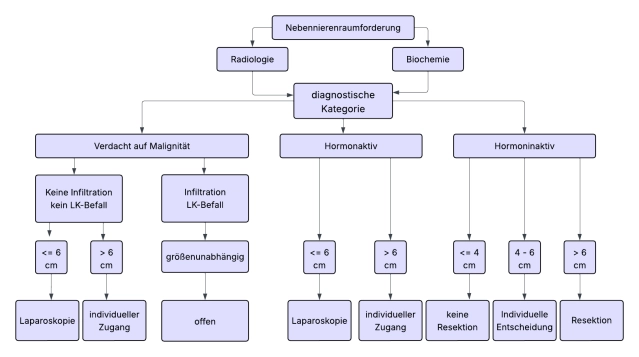
There is consensus in the guidelines that most adrenal tumors should be operated on minimally invasively when indicated. Tumor diameter >6 cm and clear indications of malignancy in preoperative imaging are considered limits of minimally invasive surgery.
For open adrenalectomy, the transabdominal or thoraco-abdominal approach is preferred, as the main indication for the open procedure is large tumors suspected of malignancy.
For suspicious adrenal tumors of 6-8 cm in size and Hounsfield units (HU) (density values of the adrenal mass) >20, an open approach (+lymphadenectomy) is preferred without preoperative evidence of malignancy.
An R0 en-bloc resection with surrounding retroperitoneal fat tissue should be aimed for, tumor capsule opening should be avoided, and at least morphologically conspicuous lymph nodes should be simultaneously removed.
In the following situations, an indication for open adrenalectomy may exist:
- Endocrine-active adrenal cortex tumors (Conn or Cushing adenomas, tumors with androgen secretion) especially with > 10 cm diameter
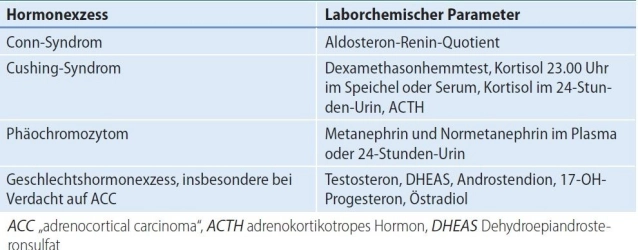
- Conn syndrome (primary hyperaldosteronism, PHA):
The most common cause of secondary hypertension is primary hyperaldosteronism.
In the case of a unilateral aldosterone-producing adenoma, unilateral adrenalectomy is indicated.
In patients with primary hyperaldosteronism (PHA) and bilateral adrenal changes, unilateral adrenalectomy may be considered if adrenal venous sampling (AVS; selective blood sampling from the adrenal veins) demonstrates clear functional lateralization. The significance of CXCR4-PET as a non-invasive alternative to AVS must be validated by further studies.
- Cushing syndrome(hypercortisolism)
A florid adrenal Cushing syndrome with classic clinical stigmata represents an indication for surgery.
Before a possible surgical removal of the adrenal tumor, ACTH independence of cortisol excess must be confirmed to prevent the procedure from being mistakenly performed, although the cause of hormone excess is, for example, pituitary due to an adenoma of the anterior pituitary (central Cushing syndrome) or paraneoplastic syndrome with ectopic ACTH secretion in tumor disease.
- Sex hormone-producing adrenal cortex tumors
An adrenocortical carcinoma is the most common cause of clinically relevant pathological androgen/estrogen secretion from the adrenal gland, adenomas are very rare.
- Conn syndrome (primary hyperaldosteronism, PHA):
- Adrenocortical carcinoma Adrenocortical carcinoma (ACC) ENSAT stage I-III Link to ENSAT classification
- At initial diagnosis, almost always > 4cm and exhibits endocrine activity in 50-80%. Typical is cortisol production or mixed hormonal production (androgens/estrogens and cortisol).
- In adrenocortical carcinoma, open adrenalectomy is the gold standard. For tumors < 6 cm without evidence of local or lymph node infiltration (ENSAT Std. I+II), minimally invasive adrenalectomy can be performed.
- A radical tumor resection with removal of the adrenal gland and all fat/connective tissue in the affected compartment without capsule opening and a lymphadenectomy, this with uncertain data situation, is recommended. A definition regarding the extent of the required lymphadenectomy is not yet available.
- Note: The rate of local and peritoneal recurrences is increased in the laparoscopic group according to current evidence. Conversion from laparoscopic to open adrenalectomy worsens overall survival.
- Pheochromocytoma (PC) Adrenal medulla tumor with catecholamine excess in cases of suspected malignancy, very large tumors, or when technical difficulties are expected.
- Approximately 1/3 of all pheochromocytoma patients are associated with a hereditary tumor syndrome. Genetic screening is an indispensable part of pheochromocytoma diagnostics. SDHB-associated tumors have a particularly high risk of malignancy and recurrence.
- Overall, 10% of PCs are malignant. Metastases are the only proof of a malignant pheochromocytoma.
- Myelolipomas do not per se constitute an indication for surgery.
Large adrenal myelolipomas (hormonally inactive benign tumors containing mature fat tissue and hematopoietic tissue) can become symptomatic through necrosis or spontaneous bleeding. In these cases, resection may be required.
- Adrenal metastases (from malignant tumors of other origins) should be removed if no further metastases are present and tumor freedom can be achieved through removal.
Note: Metastasis adrenalectomy should be performed minimally invasively, provided the metastasis can be removed in toto and without tumor cell dissemination. An open approach is reserved for the few cases where there is evidence of local infiltration or if the metastasis exceeds 6 cm.
- Large schwannomas or adult neuroblastomas