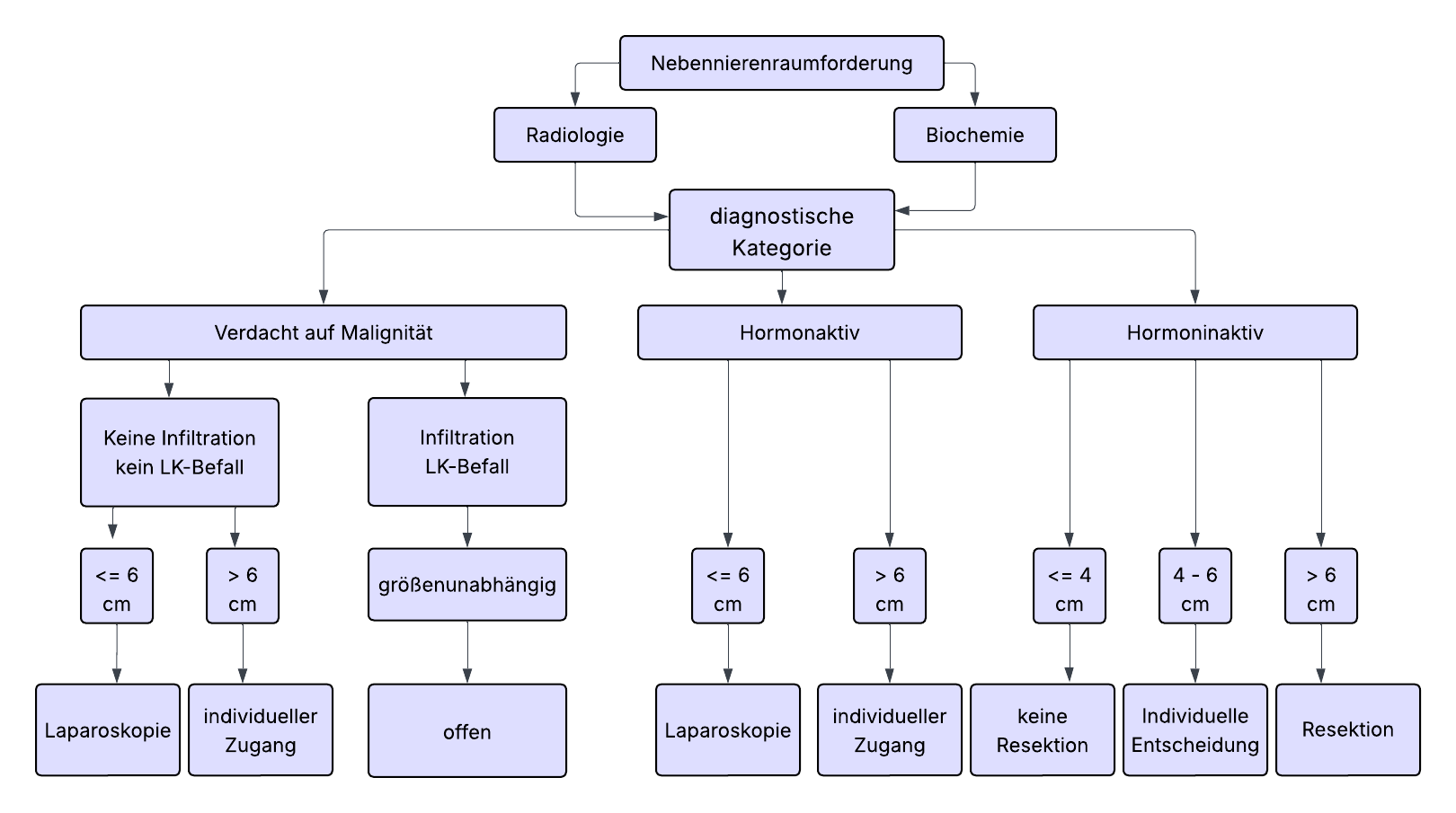
There is consensus in the guidelines that most adrenal tumors (ATs) should be operated on minimally invasively if indicated. Tumor diameter > 6 cm and clear evidence of malignancy in preoperative imaging are considered limits of minimally invasive surgery. Regarding tumor size, a 6 cm rule is often assumed, larger tumors should be excised conventionally open. In contrast, numerous studies have shown that even large adrenal tumors > 6 cm can be safely approached minimally invasively with appropriate expertise. Due to lack of evidence, the 6 cm rule cannot therefore be regarded as an absolute limit. This decision requires special information and consent of the patients. If this is present, the adrenalectomy, if the tumor can be completely removed without capsule rupture, can be performed minimally invasively regardless of tumor size.
Nevertheless, conventional adrenalectomy is generally recommended for large tumors, especially in cases of imaging suspicion of malignancy.
Laparoscopic and retroperitoneoscopic procedures are equivalent. The choice of access route depends on the experience and preference of the surgeon.
Note: Laparoscopic adrenalectomy (AE) has not changed technically significantly since its first description in 1992 and is the preferred method in many places due to the familiar access and orientation.
Partial adrenal resection (adrenal-sparing adrenalectomy) has a role in patients with Conn's syndrome, which is caused by small, usually eccentrically located tumors, and bilateral tumors, where the surgical intervention aims to preserve adrenocortical function. The aim is to preserve at least one third of an adrenal gland. The central adrenal vein does not need to be preserved. In unilateral surgery only, corticosteroid hormone production is usually completely taken over by the contralateral adrenal gland.
- Endocrine-active tumors of the adrenal cortex (Conn or Cushing adenomas, tumors with sex hormone secretion) up to 10 cm
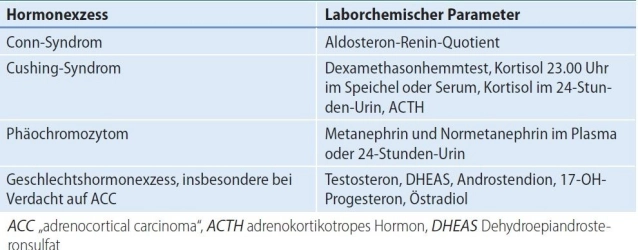
- Conn's syndrome (primary hyperaldosteronism, PHA):
The most common cause of secondary hypertension is primary hyperaldosteronism.
Screening for incidentaloma and concomitant hypertension and/or unclear hypokalemia. Morphologically, adrenal hyperplasia or one or more adenomas may be present.
In unilateral aldosterone-producing adenoma or unilateral adrenal hyperplasia, unilateral adrenalectomy is indicated. In solitary aldosterone-producing adenoma, partial removal of the affected adrenal gland can also be performed.
In patients with PHA and bilateral adrenal changes, surgery can be considered if adrenal venous sampling (AVS through selective adrenal vein blood sampling) shows functional localization.
- Cushing's syndrome(hypercortisolism)
A florid adrenal Cushing's syndrome with classic clinical stigmata represents an indication for surgery.
In subclinical Cushing's syndrome, there is only a relative indication for surgery. Here, an affected person shows biochemical cortisol excess, but clinical manifestation is absent. Comparative data on mortality or cardiovascular events of surgery versus medical therapy of comorbidities are lacking. The decision for surgery depends mainly on the age and wishes of the patient.
Before possible surgical removal of the AT, the ACTH independence of the cortisol excess must be confirmed so that the procedure is not performed erroneously, although the cause of the hormone excess is, for example, pituitary.
Exclusion of ACTH-dependent hypercortisolism (adenoma of the anterior pituitary lobe) (central Cushing's syndrome) or paraneoplastic syndrome with ectopic ACTH secretion in tumor disease.
- Sex hormone-producing adrenal cortex tumors
An adrenocortical carcinoma is the most common cause of clinically relevant, pathological androgen/estrogen secretion from the adrenal gland, adenomas are very rare.
- Conn's syndrome (primary hyperaldosteronism, PHA):
- Adrenal cortex carcinoma Adrenocortical carcinoma (ACC) up to ENSAT stage II
Link to ENSAT classification and size ≤ 6cm- At initial diagnosis almost always > 4cm and shows endocrine activity in 50-80%. Typically, there is cortisol production or mixed hormonal production (androgens/estrogens and cortisol).
- For adrenal cortex carcinoma, open adrenalectomy is the gold standard. For tumors < 6 cm without evidence of local or lymph node infiltration (ENSAT std. I+II), minimally invasive adrenalectomy can be performed.
- A radical tumor resection with removal of the adrenal gland and all fat/connective tissue in the affected compartment without capsule opening with lymphadenectomy, this in uncertain data situation, is recommended. A definition regarding the extent of the required lymphadenectomy does not yet exist.
- Note: The rate of local and peritoneal recurrences is increased in the laparoscopic group according to current evidence. Conversion from laparoscopic to open adrenalectomy worsens overall survival.
- Pheochromocytoma (PC) Adrenal marrow tumor with catecholamine excess
- About 1/3 of all pheochromocytoma patients are attributable to a hereditary tumor syndrome. Genetic screening is an indispensable part of pheochromocytoma diagnostics.
- Familial syndromic forms include multiple endocrine neoplasia type 2 (MEN 2), von Hippel-Lindau syndrome (VHL), neurofibromatosis type 1 (NF1), and germline mutations of succinate dehydrogenase subunit B and D (SDHB and SDHD).
- They differ from sporadic forms in age of onset, tumor location, and their risk of malignant degeneration.
- Syndrome-associated hereditary pheochromocytomas have a lower malignancy rate than sporadic tumors but often occur multifocally/bilaterally. The goal here is to enable a life without steroid dependence. Therefore, in this situation, a minimally invasive organ-sparing adrenal resection should be discussed.
- An important exception are SDHB-associated tumors with particularly high malignancy and recurrence risk.
- Overall, 10% of PCs are malignant. Proof of a malignant pheochromocytoma is exclusively metastases.
- Incidentalomas
| Size of the incidentaloma | Recommendation |
|---|---|
| < 4 cm | No surgery if hormonally inactive and benign imaging |
| 4 – 6 cm | Individual decision ("gray area"), weighing malignancy risk, imaging, growth, patient preference, re-CT/MRI in 6 - 12 months |
| > 6 cm | Surgery recommended, even for hormonally inactive tumors due to increased carcinoma risk (25%) |
An adrenal incidentaloma is an incidental, usually asymptomatic mass (lesion) of the adrenal gland that was not discovered due to clinical suspicion of an endocrine disease. Usually, a size of 1 cm in diameter or more is considered a relevant finding. Smaller lesions are often considered incidental findings without further need for action, unless they show abnormalities.
Note: Incidentalomas must be assessed using biochemical and radiological methods.
Only detectable infiltration of neighboring structures or detection of distant metastases are proof of malignancy. Determination of Hounsfield units (HU) can provide additional clues.
- Myelolipomas/adrenal cysts do not represent an indication for surgery. Tumor diameter is prognostically insignificant as these are benign tumors. Flank pain due to tumor size or hemorrhage in the retroperitoneum can rarely represent an indication for surgery.
- Adrenal metastases (from malignant tumors of other origins) should be removed if no other metastases are present and tumor-free status is achieved by removal.
Metastasis adrenalectomy should be performed minimally invasively, provided the metastasis can be removed in toto and without tumor cell seeding. An open approach remains reserved for the few cases where there is evidence of local infiltration or if the metastasis exceeds 6 cm.
Lymph node dissection
In enlarged lymph nodes, locoregional lymphadenectomy should be performed. The data situation regarding periadrenal/renal hilar lymphadenectomy is unsatisfactory.