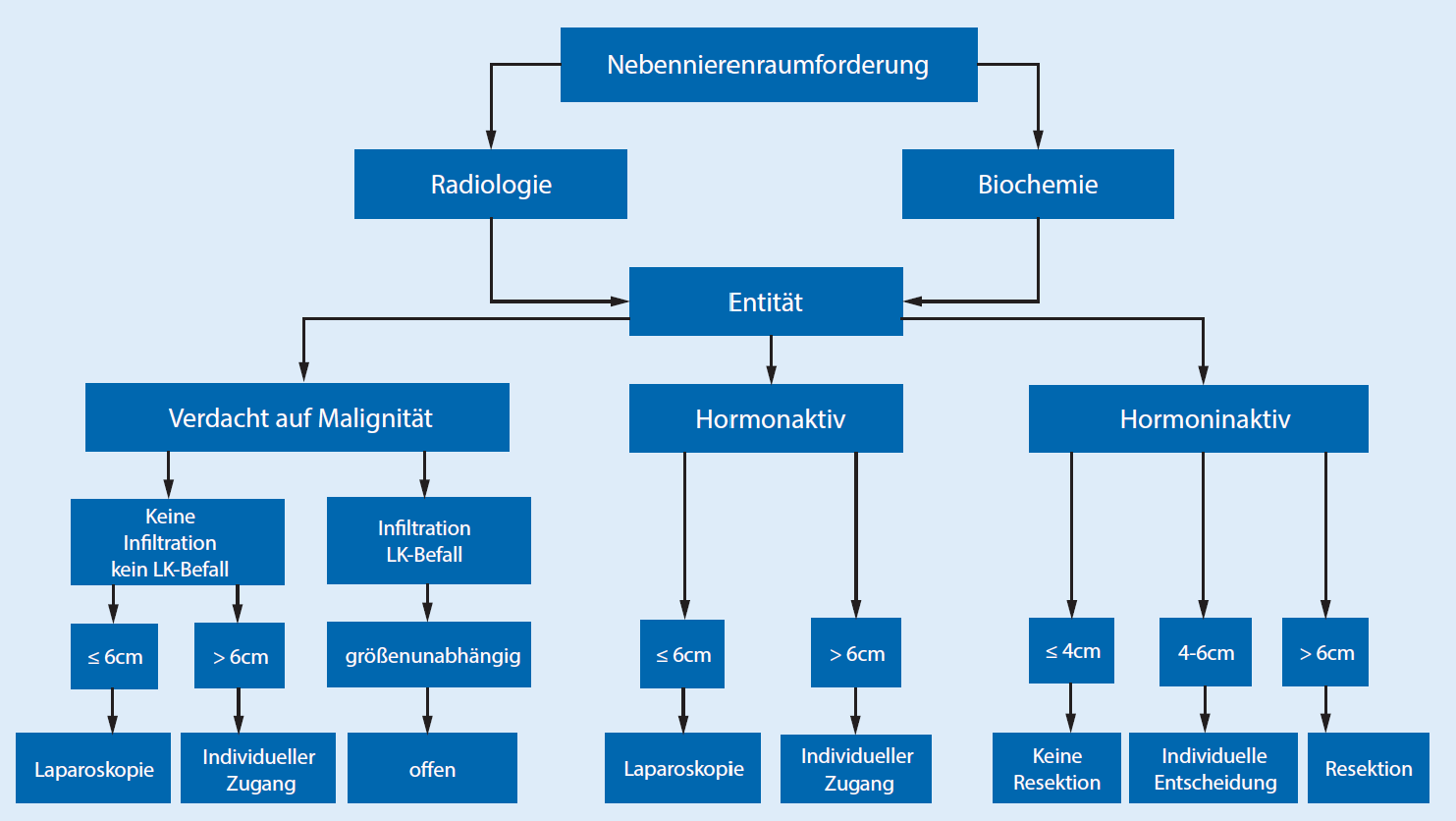
En las directrices existe consenso en que la mayoría de los tumores suprarrenales (tumores NN) deben operarse de forma mínimamente invasiva si existe una indicación dada. El diámetro del tumor > 6 cm y claros indicios de malignidad en la imagen preoperatoria se consideran límites de la cirugía mínimamente invasiva. En cuanto al tamaño del tumor, a menudo se parte de una regla de 6 cm, los tumores más grandes deben extirparse de forma convencional abierta. Por el contrario, numerosos estudios han demostrado que también en tumores suprarrenales grandes > 6 cm se puede proceder de forma mínimamente invasiva con seguridad si se cuenta con la experiencia adecuada. Debido a la falta de evidencia, la regla de 6 cm no puede considerarse un límite absoluto. Esta decisión requiere una información especial y el consentimiento de los pacientes. Si este existe, la adrenalectomía puede realizarse de forma mínimamente invasiva, independientemente del tamaño del tumor, si el tumor es completamente removible sin ruptura de la cápsula.
Sin embargo, en general, en tumores grandes, especialmente con sospecha de malignidad en la imagen, se recomienda la adrenalectomía convencional.
Los procedimientos laparoscópicos y retroperitoneoscópicos son equivalentes. La elección de la vía de acceso depende de la experiencia y la preferencia del operador.
Nota: La adrenalectomía laparoscópica (AE) se ha mantenido técnicamente sin cambios significativos desde su primera descripción en 1992 y es el método preferido en muchos lugares debido al acceso y orientación familiares.
La resección parcial de la suprarrenal (adrenalectomía preservadora de la suprarrenal) tiene un valor en pacientes con síndrome de Conn, causado por tumores pequeños generalmente excéntricos, y tumores bilaterales, en los que se busca el procedimiento quirúrgico con el objetivo de preservar la función adrenocortical. En este caso, se debe lograr la preservación de al menos un tercio de una suprarrenal. La vena suprarrenal central no necesita preservarse. En cirugía unilateral única, la producción de hormonas corticoideas es generalmente asumida completamente por la suprarrenal contralateral.
- Tumores endocrino-activos de la corteza suprarrenal (adenomas de Conn o Cushing, tumores con secreción de hormonas sexuales) hasta 10 cm
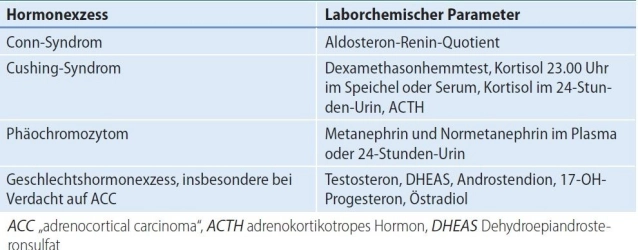
- Síndrome de Conn (hiperaldosteronismo primario, PHA):
La causa más frecuente de hipertensión secundaria es el hiperaldosteronismo primario.
Screening en incidentalomas y hipertensión simultánea y/o hipopotasemia inexplicada. Morfológicamente puede haber una hiperplasia de las suprarrenales o uno o varios adenomas.
En un adenoma productor de aldosterona unilateral o una hiperplasia adrenal unilateral, está indicada la adrenalectomía unilateral. En el adenoma productor de aldosterona solitario, también se puede realizar una extirpación parcial de la suprarrenal afectada.
En pacientes con PHA y alteraciones bilaterales de las suprarrenales, se puede considerar una operación si el muestreo venoso adrenal (AVS mediante extracción selectiva de sangre de las venas suprarrenales) muestra una localización funcional.
- Síndrome de Cushing(hipercortisolismo)
Un síndrome de Cushing adrenal florido con estigmas clínicos clásicos representa una indicación quirúrgica.
En el síndrome de Cushing subclínico solo existe una indicación quirúrgica relativa. En este caso, el afectado muestra bioquímicamente un exceso de cortisol, pero falta la manifestación clínica. Faltan datos comparativos sobre la mortalidad o eventos cardiovasculares de la operación frente a una terapia medicamentosa de las comorbilidades. La decisión quirúrgica depende principalmente de la edad y el deseo del paciente.
Antes de una posible extirpación quirúrgica del tumor NN, debe confirmarse la independencia de ACTH del exceso de cortisol, para que la intervención no se realice erróneamente, aunque la causa del exceso hormonal sea, por ejemplo, de origen hipofisario.
Exclusión de hipercortisolismo dependiente de ACTH (adenoma del lóbulo anterior de la hipófisis) (síndrome de Cushing central) o síndrome paraneoplásico con secreción ectópica de ACTH en enfermedad tumoral.
- Tumores de la corteza suprarrenal productores de hormonas sexuales
Un carcinoma adrenocortical es la causa más frecuente de una secreción patológica clínicamente relevante de andrógenos/estrógenos de la suprarrenal, los adenomas son muy raros.
- Síndrome de Conn (hiperaldosteronismo primario, PHA):
- Carcinoma de la corteza suprarrenal Carcinoma adrenocortical (ACC) hasta el estadio ENSAT II
Enlace a la clasificación ENSAT y tamaño ≤ 6cm- En el diagnóstico inicial casi siempre > 4cm y presenta en 50-80% una actividad endocrina. Típica es una producción de cortisol o una producción hormonal mixta (andrógenos/estrógenos y cortisol).
- En el carcinoma de la corteza suprarrenal, la adrenalectomía abierta es el estándar de oro. En tumores < 6 cm sin indicios de infiltración local o linfática (ENSAT est. I+II) se puede realizar una adrenalectomía mínimamente invasiva.
- Se recomienda una resección tumoral radical con extirpación de la suprarrenal y de todo el tejido graso/conectivo en el compartimento afectado sin apertura de la cápsula con linfadenectomía, esta en caso de datos inciertos. No existe aún una definición respecto a la extensión de la linfadenectomía requerida.
- Nota: La tasa de recidivas locales y peritoneales está aumentada en el grupo laparoscópico según la evidencia actual. Una conversión de adrenalectomía laparoscópica a abierta empeora la supervivencia global.
- Feocromocitoma (PC) Tumor de la médula suprarrenal con exceso de catecolaminas
- Aproximadamente 1/3 de todos los pacientes con feocromocitoma se asocian a un síndrome tumoral hereditario. El screening genético es una parte indispensable del diagnóstico de feocromocitoma.
- Entre las formas sindrómicas familiares se incluyen la neoplasia endocrina múltiple tipo 2 (MEN 2), el síndrome de von Hippel-Lindau (VHL), la neurofibromatosis tipo 1 (NF1) y las mutaciones germinales de la subunidad B y D de la succinato deshidrogenasa (SDHB y SDHD).
- Se diferencian de las formas esporádicas por la edad de aparición, la localización del tumor y su riesgo de degeneración maligna.
- Los feocromocitomas hereditarios asociados a síndromes tienen una tasa de malignidad más baja que los tumores esporádicos, pero a menudo ocurren de forma multifocal/bilateral. El objetivo aquí es permitir una vida sin dependencia de esteroides. Por lo tanto, en esta situación se debe discutir una resección suprarrenal mínimamente invasiva preservadora de órgano.
- Una excepción importante son los tumores asociados a SDHB con riesgo particularmente alto de malignidad y recidiva.
- En total, el 10 % de los PC son malignos. La prueba de un feocromocitoma maligno son exclusivamente las metástasis.
- Incidentalomas
| Tamaño del incidentaloma | Recomendación |
|---|---|
| < 4 cm | Sin OP, si hormonalmente inactivo y con imagen benigna |
| 4 – 6 cm | Decisión individual ("zona gris"), ponderación del riesgo de malignidad, imagen, crecimiento, deseo del paciente, Re-CT/MRT en 6 - 12 meses |
| > 6 cm | OP recomendada, también en tumores hormonalmente inactivos debido al riesgo aumentado de carcinoma (25%) |
Un incidentaloma suprarrenal es una masa ocupante de espacio (RF) suprarrenal accidental, generalmente asintomática, que no se descubrió debido a una sospecha clínica de una enfermedad endocrina. Generalmente, un tamaño a partir de 1 cm de diámetro se considera un hallazgo relevante. Las lesiones más pequeñas a menudo se consideran hallazgos incidentales sin necesidad de acción adicional, a menos que muestren anomalías.
Nota: Los incidentalomas deben evaluarse mediante métodos bioquímicos y radiológicos.
Solo la infiltración reconocible de estructuras vecinas o la detección de metástasis distantes son prueba de malignidad. La determinación de las unidades Hounsfield (HU) puede proporcionar indicios adicionales.
- Mielolipomas/quistes suprarrenales no representan una indicación quirúrgica. El diámetro del tumor es pronósticamente insignificante, ya que se trata de tumores benignos. Dolor en el flanco debido al tamaño del tumor o una hemorragia en el retroperitoneo pueden raramente representar una indicación quirúrgica.
- Metástasis suprarrenales (de tumores malignos de otro origen) deben extirparse si no hay otras metástasis y se logra la ausencia de tumor mediante la extirpación.
La adrenalectomía de metástasis debe realizarse de forma mínimamente invasiva, siempre que la metástasis se pueda extirpar in toto y sin diseminación de células tumorales. Un procedimiento abierto se reserva para los pocos casos en los que hay indicios de infiltración local o si la metástasis supera los 6 cm.
Disección de ganglios linfáticos
En ganglios linfáticos agrandados se debe realizar una linfadenectomía locorregional. La situación de los datos respecto a una linfadenectomía periadrenal/renal-hilar es insatisfactoria.